
Py-ChemShell 2023 released
We are delighted to announce the release of Py-ChemShell 2023 (v23.0), the latest release of the Python-based version of ChemShell.
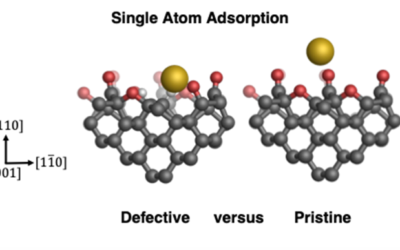
Stability of single gold atoms on defective and doped diamond surfaces
QM/MM calculations show that surface defects (vacancies and dopants) promote the stability of single gold atoms on diamond.
ChemShell featured on PCCP cover
Our recent Perspective article on ChemShell is highlighted on the cover of the latest issue of Phys. Chem. Chem. Phys.
ChemShell developer vacancy at STFC [closed]
An open ended position is available at STFC to join the team developing ChemShell.
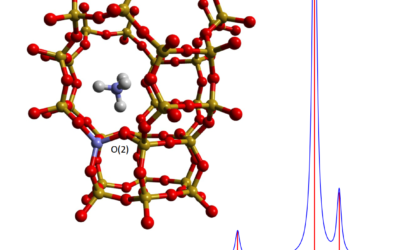
Computational infrared and Raman spectra by hybrid QM/MM in ChemShell
New functionality in ChemShell enables the calculation of QM/MM infrared and Raman spectra for molecular and catalytic materials systems.

Multiscale QM/MM modelling of catalytic systems with ChemShell
A new PCCP Perspective is available covering recent ChemShell development and applications work for modelling catalysis.
PhD studentship on high temperature superconductivity [closed]
A PhD studentship is available at University College London for a project to model high temperature superconductors using ChemShell.
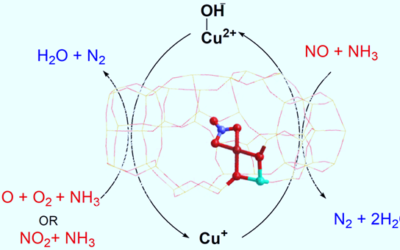
Influence of solvent on selective catalytic reduction of nitrogen oxides with ammonia over Cu-CHA zeolite
The NH3-SCR reaction mechanism on Cu-CHA has been studied using QM/MM calculations and DRIFTS spectroscopy to investigate solvent effects.
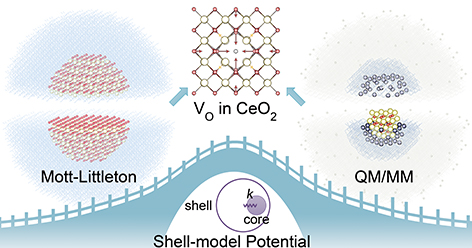
QM/MM calculations assist the development of a robust shell-model interatomic potential for CeO2
A novel strategy that employs accurate ionic polarisabilities, defect structures, and formation energies calculated by the QM/MM ...
Exascale materials modelling vacancy [closed]
We are looking for a postdoc to join the Computational Chemistry Group at STFC Daresbury Laboratory to work on multiscale materials ...
Py-ChemShell 2021 released
We are delighted to announce the release of Py-ChemShell 2021 (v21.0), the third beta release of the Python-based version of ChemShell.
Geometry optimisation based on machine learning
Johannes Kästner's group at the University of Stuttgart have extended DL-FIND to use Gaussian-process regression (GPR) to search for ...
Py-ChemShell 2020 released
We are delighted to announce the release of Py-ChemShell 2020 (v20.0), the second beta release of the Python-based version of ChemShell.
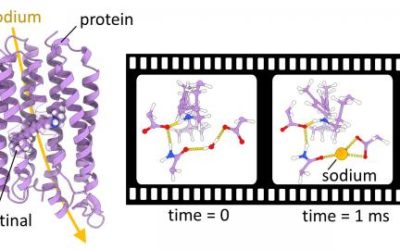
QM/MM simulations help to resolve the sodium pumping mechanism of a light-driven ion transporter
Krokinobacter eikastus rhodopsin 2 (KR2) is a light-driven sodium pump that actively transports small cations across cellular ...
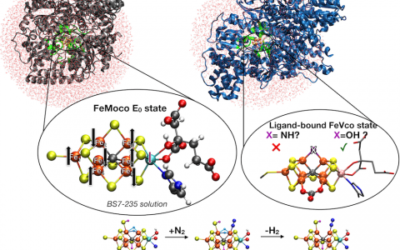
QM/MM studies of molybdenum and vanadium nitrogenases
The iron-molybdenum (FeMoco) and iron-vanadium cofactors (FeVco) in the molybdenum/vanadium nitrogenase enzymes catalyze the reduction ...
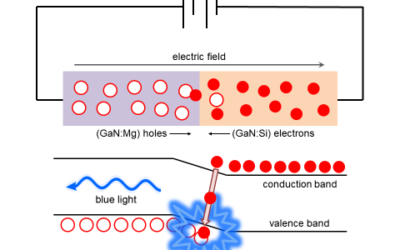
Defects in the wide gap semiconductor GaN
GaN is a wide gap semiconductor that is a crucial component of blue light emitting diodes (LEDs), which are essential for solid state ...

In memory of Walter Thiel
We were deeply shocked and saddened to hear that our colleague, friend and mentor Prof. Walter Thiel passed away last month.
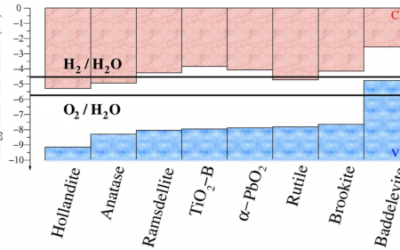
Determining the energy band alignment between different TiO2 polymorphs
TiO2 is a key material for photocatalytic water splitting, where it has been found that samples composed of mixtures of the anatase ...
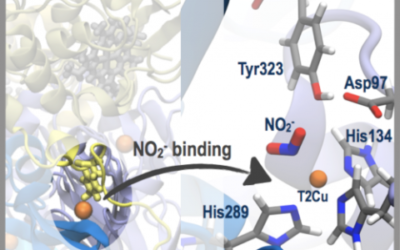
Nitrite binding in three-domain haem-Cu nitrite reductase
Copper-containing nitrite reductase enzymes (CuNiRs) play a key role in the global nitrogen cycle by reducing nitrite (NO2−) to nitric ...
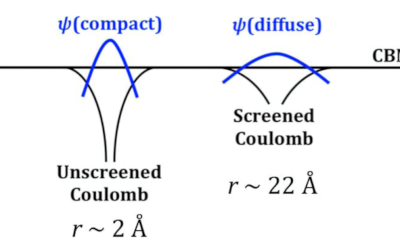
Understanding how oxygen vacancies affect conductivity in transparent conducting oxides
The role of oxygen vacancies in the transparent conducting oxides (TCOs) In2O3, SnO2 and ZnO has remained controversial, with some ...
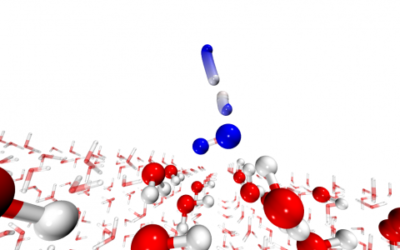
Water formation in the interstellar medium
Chemical reactions in the interstellar medium occur at low temperature, often on ice surfaces. T
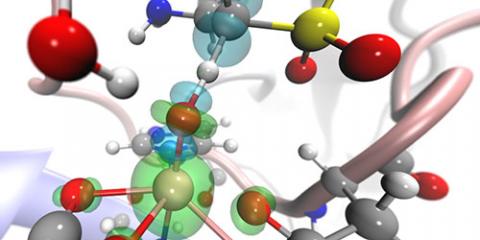
Unravelling of the reaction mechanism and tunnelling contributions in Taurine Dioxygenase
The biochemical turnover of taurin is catalysed by taurine dioxygenase, aided by molecular oxygen and alpha-ketogluterate.
Highlight your research on this site!
If you've used ChemShell in published research, we would like to highlight it on this page and on our Twitter feed to help publicise ...
Py-ChemShell 2019 released
We are pleased to announce Py-ChemShell 2019 (v19.0), the first beta release of the Python-based version of ChemShell.
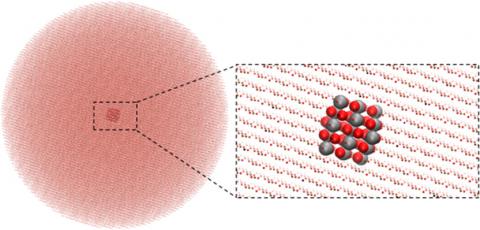
ChemShell redevelopment work published
The project to redevelop ChemShell as a python-based program has been published in the Journal of Chemical Theory and Computation.